
Test prenatale non invasivo (da sangue materno)
Durante la gravidanza, frammenti di DNA libero del bambino circolano nel sangue materno. Il DNA fetale è rilevabile a partire dalla 5a settimana di gestazione e la sua concentrazione aumenta durante le settimane successive. La quantità di DNA fetale presente nel circolo sanguigno materno a partire dalla 10a settimana di gestazione (12a settimana in caso di gravidanza gemellare) è sufficiente per eseguire il test e garantire risultati accurati.
Dal campione di sangue materno viene estratto e purificato il DNA. Successivamente il DNA viene analizzato mediante sequenziamento massivo in parallelo dell’intero genoma utilizzando la tecnologia NGS (Next Generation Sequencing). Ciò consente di avere lo screening più completo. I dati di sequenziamento sono accuratamente analizzati da sistemi di bioinformatica avanzati, per fornire referti chiari e affidabili.
Le anomalie cromosomiche numeriche (aneuploidie) si verificano quando un individuo presenta
un cromosoma in più rispetto all’usuale coppia di cromosomi (trisomia), oppure quando in una coppia di cromosomi ne manca uno (monosomia).
TRISOMIA 21 (Sindrome di Down)
È un’anomalia cromosomica caratterizzata dalla presenza di un cromosoma 21 (completo o di parte di esso) in sovrannumero. Le principali malformazioni e complicazioni comprendono le cardiopatie, le malformazioni intestinali, la cataratta congenita e la bassa statura.
TRISOMIA 18 (Sindrome di Edwards)
È un’anomalia cromosomica caratterizzata dalla presenza di un cromosoma 18 in sovrannumero
associata a ritardo della crescita, dolicocefalia, facies caratteristica, anomalie degli arti e malformazioni viscerali.
TRISOMIA 13 (Sindrome di Patau)
È un’anomalia cromosomica causata dalla presenza di un cromosoma 13 in sovrannumero ed è caratterizzata da malformazioni cerebrali (oloprosencefalia), dismorfismi facciali, anomalie oculari, polidattilia postassiale, malformazioni viscerali (cardiopatia) e grave ritardo psicomotorio.
Causano la perdita o il guadagno di materiale genetico. Le principali anomalie strutturali individuabili col NIPT I livello sono le delezioni e le duplicazioni.
ANEUPLOIDIE DEI CROMOSOMI SESSUALI
I cromosomi X e Y determinano il sesso del bambino. Le principali combinazioni anomale osservate sono XXX, XYY (Sindrome di Jacobs), XXY (Sindrome di Klinefelter), e monosomia X (Sindrome di Turner). Gli individui affetti da tali sindromi possono presentare disturbi fisici e comportamentali la cui gravità può variare sensibilmente da soggetto a soggetto.
Le anomalie strutturali includono la mancanza o la delezione di una porzione di cromosoma.
MICRODELEZIONI
Si manifestano quando un segmento di un cromosoma presenta una piccola delezione. Le sindromi da microdelezione sono delle patologie clinicamente riconoscibili, caratterizzate da un complesso fenotipo clinico e comportamentale ed includono la sindrome di DiGeorge, la sindrome del Cri-du-Chat, la sindrome di Prader-Willi, la sindrome di Angelman, la sindrome di Wolf Hirshhorn, la sindrome da delezione1p36, Sindrome di Jacobsen, Sindrome di Langer-Giedion, Sindrome di Smith-Magenis e Sindrome da Brachidattilia-deficit cognitivo, non sono più una fatalità.
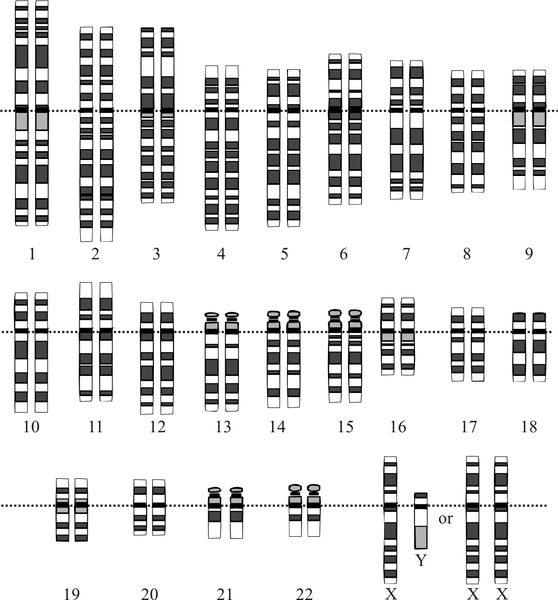
Analisi prenatale del cariotipo fetale su sangue materno
Il test prevede, oltre a quanto descritto precedentemente, anche l’opzione di un approfondimento diagnostico di secondo livello, che consente di individuare la presenza nel feto di alterazioni cromosomiche strutturali ed alcune comuni sindromi da microdelezione/ microduplicazione.
L’altissimo grado di affidabilità riduce sensibilmente la possibilità di essere inutilmente sottoposte a rischio di indagini diagnostiche invasive (Amniocentesi e Villocentesi).
Anche tale prelievo può essere eseguito a partire dalla decima settimana di gestazione.
Test Dna fetale NIPT di massimo screening
Sequenziamento massivo parallelo dell’intero genoma -MPS- da DNA Fetale Circolante. E’ il primo test prenatale non invasivo in Europa in grado di rilevare le patologie mendeliane a trasmissione autosomica recessiva e/o a trasmissione autosomica dominante
– Fibrosi Cistica CFTR
– Sordità ereditaria tipo 1A CX26 (GJB6)
– Sordità ereditaria tipo 1B CX26 (GJB6)
– Beta Talassemia HBB
– Anemia falciforme HBB
– Fenilchetonuria PAH
e Sindromi:
– Sindrome di Gaucher GBA
– Sindrome Dubowitz LIG4-NSUN2
– Sindrome di Richner-Hanhart TAT
– Sindrome di Sjögren-Larsson ALDH3A2
– Sindrome di Costello HRAS
– Sindrome di Tay-Sachs HEXA
– Sindrome di PKAN PANK2
– Sindrome della Tripla-H SLC25A15
– Sindrome di Coffin-Lowry RPS6KA3
A queste si possono aggiungere altre patologie da investigare: ulteriori malattie sindromiche, Patologie scheletriche, Craniosinostosi
Le mutazioni individuate in questi geni possono insorgere in modo casuale nel feto. Tali mutazioni, non sempre sono rilevabili nei genitori con i test di screening pre-concezionali, poichè possono essere anche non ereditarie. La presenza di mutazioni in uno dei geni investigati può causare nel bambino displasie scheletriche, difetti cardiaci, anomalie congenite multiple, autismo, epilessia e/o deficit intellettivi.
I test possono essere eseguiti a partire dalla 10° settimana di gestazione, in caso di gravidanza singola, oppure dalla 12° settimana in caso di gravidanza gemellare.Non esiste un termine entro la quale eseguire i test, anche se dal punto di vista clinico è preferibile eseguirlo, entro la 17° settimana, per poter svolgere un amniocentesi di conferma in caso di esito positivo.
Non è necessario il digiuno.
Sarà nostra cura aiutare la paziente nella comprensione del referto fornendo un’appropriata interpretazione.